Research
1. Electrochemical Reaction
1.1 Carbon Dioxide Reduction
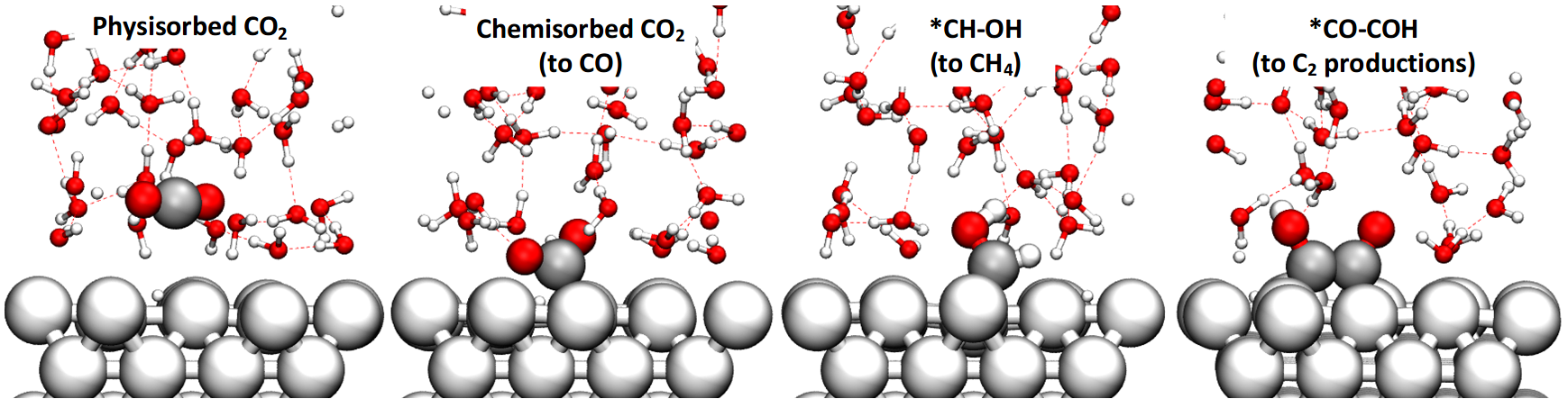
A critical step towards rational design of new catalysts that achieve selective and efficient reduction of carbon dioxide to specific hydrocarbons and oxygenates is to determine the detailed reaction mechanism including kinetics and product selectivity as a function of pH and applied potential for known systems. To accomplish this we apply ab initio molecular dynamics free energy calculation for the water/Cu system with explicit solvent to determine the kinetics and pathways for major products (ethylene and methane) and minor products (ethanol, glyoxal, glycoladehyde, ethylene glycol, acetaldehyde, ethane and methanol).
Related publications:
Surface Ligand Promotion of CO2 Reduction Ultrahigh Mass Activity for CO2 Reduction Nature of Active Sites for CO Reduction on Cu Nanoparticles Predicted Structures of Active Sites for CO Reduction Full Atomistic Reaction Mechanism for CO Reduction on Cu(100) Reaction Mechanisms for Electrochemical Reduction of CO2 Free-Energy Barriers for CO Reduction on Cu(100)1.2 Oxygen Reduction Reaction
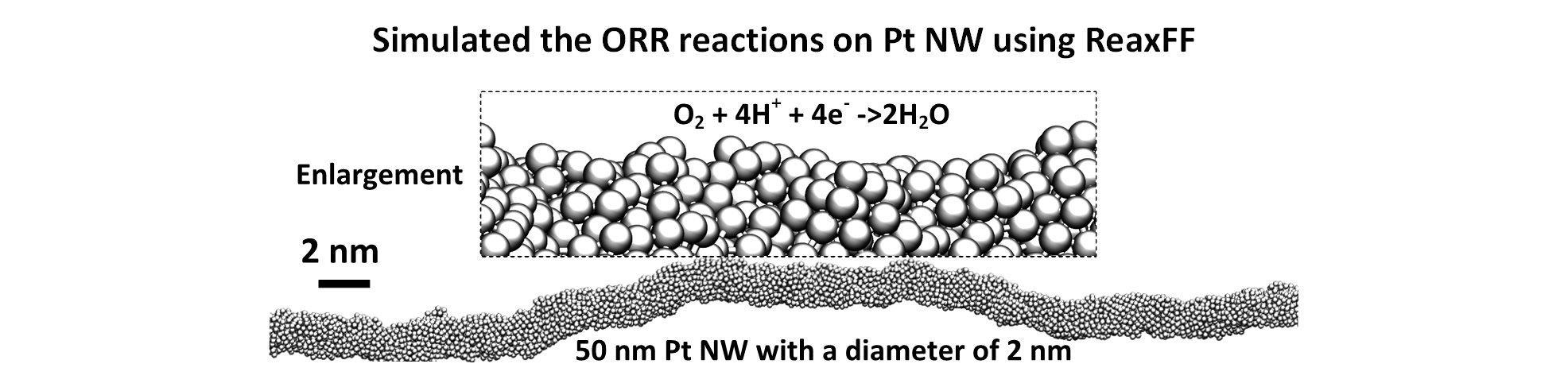
The sluggish Oxygen Reduction Reaction (ORR) is a major impediment to economical use of fuel cells in transportation. However, the reaction mechanism of ORR is still far from clear. Here, we explored the full ORR reaction mechanism for Pt(111) based on ab initio molecular dynamics free energy calculations including explicit water.
Related publications:
Mechanism and Kinetics of Electrocatalytic ORR Ultrafine Jagged Pt Nanowires for ORR2. Battery Interface/Interphase
Solid Electrolyte Interface2.1 Solid Electrolyte Interface
3. Advanced Methods
Accelerated Molecular Dynamics3.1 Accelerated Molecular Dynamics
We develop the methodology for dramatically accelerating the ReaxFF reactive force field based reactive molecular dynamics simulations through use of the bond boost concept, which we validate for describing hydrogen combustion.
Related publications:
Adaptive Accelerated ReaxFF Reactive Dynamics